Research Interests
I am interested in genomic sequence related algorithm development, such as writing tools for genomic variation detection and representation.
Education and Qualifications
Ph.D. Program in Bioinformatics: Boston College, MA, USA
Sep. 2009 - Present
Ph.D. Program Courses Taken: Biological Chemistry, Advanced Genetics, Advanced Cell Biology, Eukaryotic Gene Expression, Viruses, Genes and Evolution, Graduate Bioinformatics, Algorithms/Computational Biology
Current Projects
SIMD Smith-Waterman C/C++ Library, profile Hidden Markov Model based genomic variation detector
Master of Science in Genomics and Pathway Biology
Division of Pathway Medicine, The University of Edinburgh, UK
Sep. 2006 - Aug. 2007
Final Mark: 73/100 A, Distinction
Graduate Thesis: “The integration of the dynamic pathway information into the Medical Microbiology Database and the improvement of the Boolean network modeling method”.
M.Sc. Program Courses Taken: Bioinformatics
Courses Taken at the Graduate University of Chinese Academy of Science (GUCAS) as employee education of Beijing Genomics Institute, P. R. China
Sep. 2005 - Jun. 2006
Molecular Biology, Biological Statistics
Bachelor of Science in Computer Science
Beijing Normal University, P.R. China
Sep. 2000 - Aug. 2004
GPA: 3.4/4.0 Undergraduate Thesis “DNA Sequencing and Human Leukocyte Antigen (HLA) Matching Software” (completed in the Beijing Genomics Institute) was awarded Excellent Graduation Thesis (95/100), 2004
Awarded Third Prize Scholarship, 2001
Employment History
Teaching Assistant, Boston College, USA
Sep. 2009 - May 2010, Sep. 2012 - Dec. 2012
Courses taught: Introduction to Molecular and Cell Biology, Anatomy and Physiology Lab, Graduate Bioinformatics
Computational Biologist, Wellcome Trust Sanger Institute, UK
Nov. 2007 - Aug. 2009
Group: Human Evolution, Group leader: Chris Tyler-Smith
Main work: Coalescence history analysis based on the SNP density distribution of 6 whole genome sequences from YRI, CEU and CHB populations (gotten from the 1000 Genome Project).
Other work: Facilitate the data process for other projects in our group. Scientific Programmer and Database Curator European Bioinformatics Institute, UK Aug. 2007- Nov. 2007
The Microarray Informatics Team, Group leader: Alvis Brazma Main
Work: Manually curate data for the Array Expression Database. Used Perl and DBI SQLite to construct a curation management system to facilitate the manual curation.
Scientific Programmer (Visiting work) Wellcome Trust Sanger Institute
Apr. 2007
Group: Signal Transduction in C. elegans, Group leader: Andrew Fraster
Main work: Use Perl to calculate the distance between a/each pair of terms of C. elegans’ genes in the Gene Ontology graph
Beijing Genomics Institute (BGI), Chinese Academy of Science, Research Assistant
Jun. 2004 - Aug. 2006
Group: Algorithm Development Team, Group leader: Heng Li
Main work: Re-annotate the rice genome. Contribute to the development of hyterozygote detection software for capillary reads. Document the data processing pipeline of HLA typing. Sequence genes of Salmon.
Other work: Help to establish the employee training and external training systems of BGI. Work as teaching assistant of the graduate course “An Introduction of Bioinformatics” in GUCAS.
Current Projects
1) SIMD Smith-Waterman (SSW) C/C++ Library
SSW is a fast implementation of the Smith-Waterman (SW) algorithm, which uses the Single-Instruction Multiple-Data (SIMD) instructions to parallelize the algorithm within one processor. SSW library provides an API that can be flexibly used by programs written in C, C++ and other languages, even in multithreaded way. We also provide a demo software that can do protein and genome alignment directly. Current version of our implementation is ~50 times faster than an ordinary Smith-Waterman. It can return the Smith-Waterman score, alignment location and trace back path (cigar) of the optimal alignment accurately; and return the sub-optimal alignment score and location heuristically.
License: MIT
Features:
-
Efficient: ~50 times faster and ~100 times less memory usage than an ordinary Smith-Waterman
-
Accurate results and more: This library always gives the accurate optimal alignment results exactly the same as an ordinary Smith-Waterman. A suboptimal alignment is also returned.
-
Feather weight and easy usage: 1) Copy ssw.h and ssw.c fromhttps://github.com/mengyao/Complete-Striped-Smith-Waterman-Library and past them into your source code folder. 2) #include “ssw.h” where you would like to use the API 3) Compile with your own codes.
-
example.c and example.cpp are two simple examples to show how to use the API in C and C++ style respectively.
-
Robust demo software is provided for you to play with whatever way you like.
-
Double layer parallelization: 1) SIMD parallelization realizes parallel pairwise alignment within each processor. 2) Multithreaded usage of SSW realizes running alignments on multiple processors simultaneously.
See more at: https://github.com/mengyao/Complete-Striped-Smith-Waterman-Library
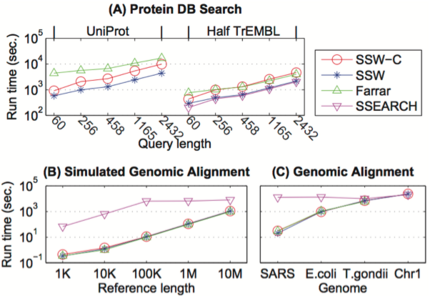
Pictured Above: Running time for different SW implementations. Log-scaled running time is shown on the y-axis for SSW without (blue) and with (red) detailed alignment, Farrar’s implementation (green) and SSEARCH (pink). (A) Running times are shown for searching protein sequences against the full UniProt database (left) and one half of the TrEMBL database (right). All SW implementations used the BLOSUM50 scoring matrix with gap open penalty -12 and extension penalty -2. (B) The running time of aligning 1,000 simulated Illumina reads to human reference sequences of various lengths; and (C) of aligning 1,000 real sequencing reads to various microorganism genomes and the human chromosome 1 are shown. Farrar’s implementation cannot handle long sequences as human chromosome 1, so its corresponding running time is not shown here.
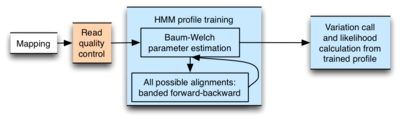
2) Profile Hidden Markov Model based genomic variation detector
A variety of algorithms have been developed to detect SNPs, insertions and deletions (INDELs), and other complex genomic variations using next- generation sequencing data. However, INDEL and other complex variation detections still face significant problems because of sequencing and alignment uncertainties. Moreover, most of the existing algorithms are designed for high-quality short reads whereas upcoming technologies tend to produce longer reads at the cost of an increased sequencing error rate. This leads to more challenging variation detection.
We are therefore developing a genomic variation caller (PHV) based on Profile Hidden Markov Model (PHMM), which jointly models sequencing and alignment uncertainties that are usually separately modeled by other callers. While this tool will show its greater advantage for more error prone data, it also demonstrates an improvement in variation detection when deployed on currently available short read technology data.
Other variation detectors’ pipeline:

PHV’s pipeline: